We use of AAV-based gene therapy strategies to treat monogenic inherited rare diseases affecting the Central Nervous System, by expressing a correct copy of the mutated gene responsible for the disease. In particular, we are currently working on Spastic Paraplegia Type 52 (SPG52), GNB1 Encephalopathy, and ADSL Deficiency. In all cases, we closely collaborate with patients and Patient’s Associations.
Spastic Paraplegia Type 52 (SPG52)
Spastic paraplegia 52 (SPG52) is a progressing neurodegenerative disorder that generally presents with global developmental delay, moderate to severe intellectual disability, impaired/absent speech, small head size (microcephaly), seizures, and progressive motor symptoms. SPG52 is an autosomal recessive ultrarare disease (less than 100 cases reported worldwide), caused by mutations in the AP4s1 gene. This gene encodes the sigma subunit of the AP4 complex, which plays important roles in the secretory and endocytic pathways by mediating vesicle formation and sorting of integral membrane proteins.
Because there is not treatment available, we are developing a gene therapy strategy using AAV vectors carrying a correct copy of the AP4s1 gene, in both, a transgenic animal model for SPG52 that we have generated in our lab, and 2D and 3D cultures of iPSC-derived neurons from patients. See following VIDEO for explanation.
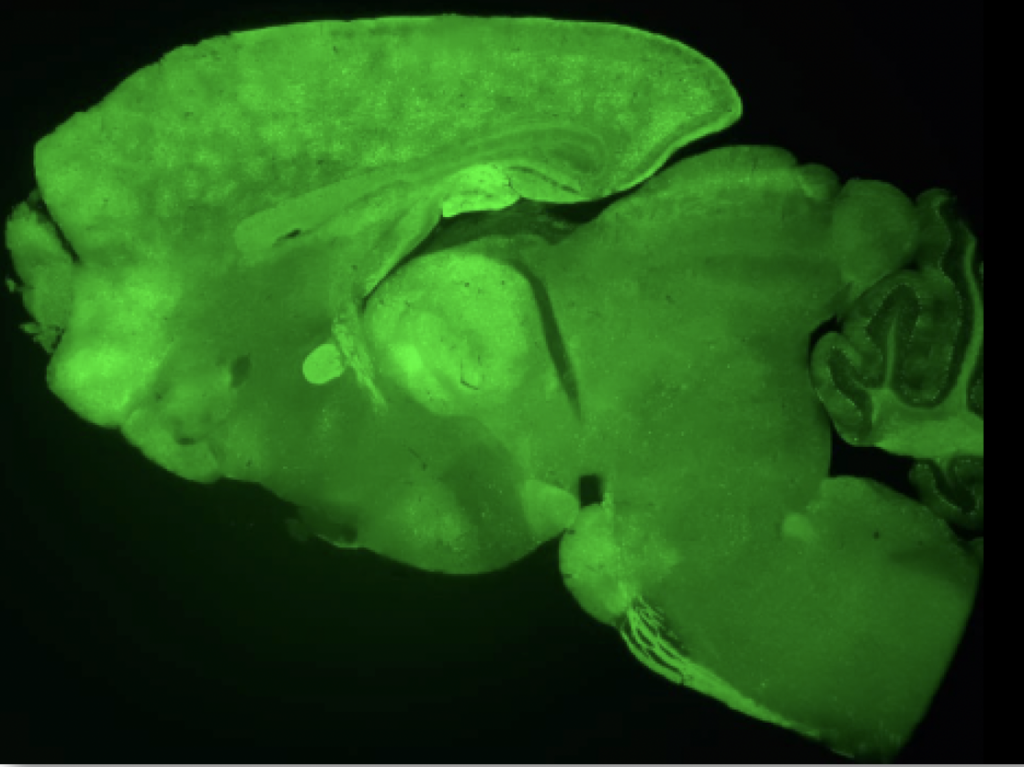

The Association «La Lucha de Abril», a patient’s association created by familiars of patients, together with a microfunding campaing at the Universitat Autònoma de Barcelona are financing the research of this disease in our group.
GNB1 Encephalopathy
GNB1 encephalopathy is a severe neurodevelopmental disorder characterized by global developmental delay, speech deficits, mobility problems, intellectual disability, and seizures. GNB1 encephalopathy is an autosomic dominant ultrarare disease (with fewer than 100 cases reported worldwide), caused by mutations in the GNB1 gene. This gene encodes the Gβ1 subunit of G proteins, central mediators of G protein-coupled receptor signaling that participate in numerous biochemical processes and pathways.
Regrettably, there is not treatment available and therefore, it is necessary to develop new treatments using cutting-edge technologies such as AAV-based gene therapy carrying a silence-and-repair expression cassette, using as models 2D and 3D cultures of iPSC-derived neurons from patients, as well as transgenic animal model for GNB1.
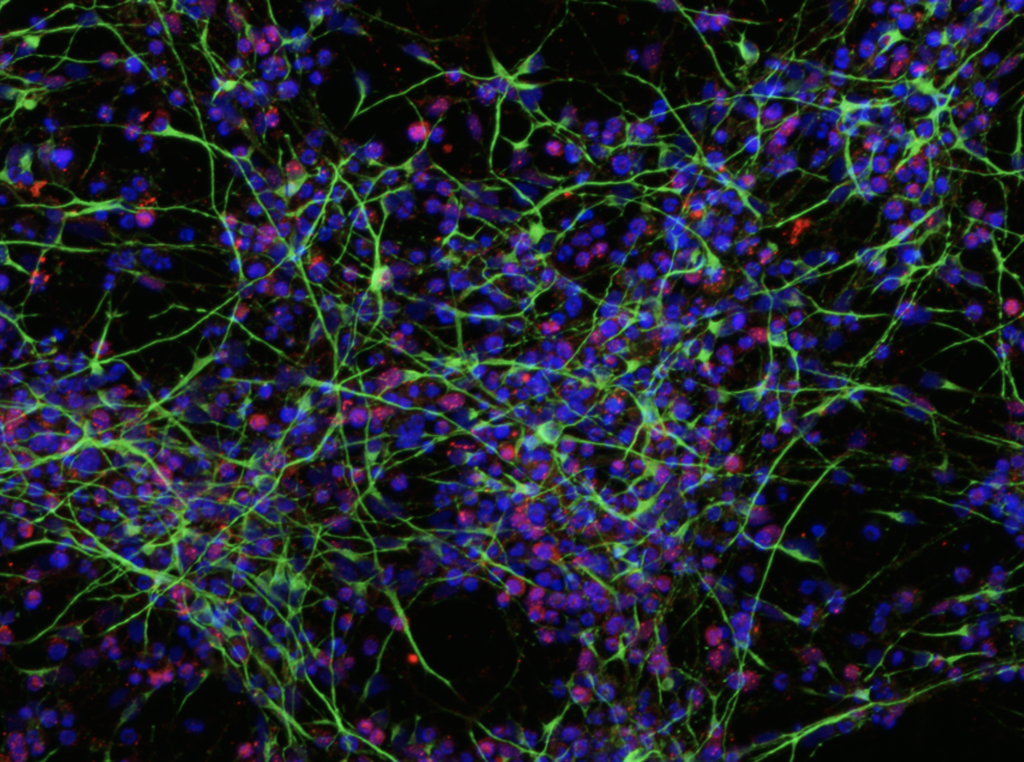
This research started thanks to the effort of the Fundacion «Gen Rebelde», a patient’s association created by familiars of GNB1 patients. In the last months other GNB1 patient’s associations such as «Asociación GNB1 España», and «GNB1 Advocacy Group» (USA), are also participating in financing the research of this disease. In addition, these initiatives are complemented through a GNB1 microfunding campaing at the Vall d’Hebron Hospital.
ADSL Deficiency
ADSL deficiency is a neurological disorder characterized by severe psychomotor delay, weak muscle tone (hypotonia), and many affected infants may develop microcephaly, recurrent seizures, and autistic traits. This autosomal recessive ultrarare disease (less than 100 cases reported worldwide) arises from mutations in the ADSL gene. This gene encodes for adenylosuccinate lyase, which performs two steps in the synthesis of purine nucleotides. These nucleotides are building blocks of DNA, RNA, and molecules such as ATP that serve as energy sources in the cell.
Unfortunately, there is not treatment available. To address it, we have just started a new gene therapy research strategy using AAV vectors carrying a correct copy of the ADSL gene. At the moment we are generating both, a transgenic animal model for ADSL Deficiency and iPSC-derived neurons from patients.
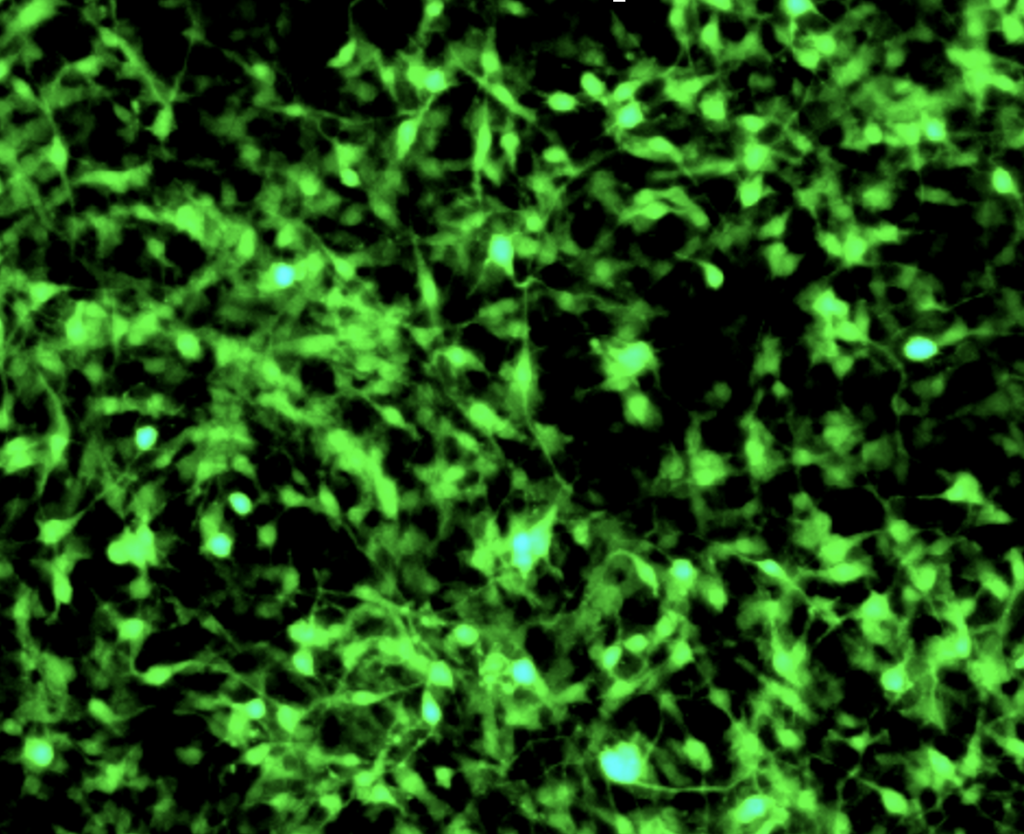

The Association «ANTIAN», a patient’s association created by familiars of patients, together with a microfunding campaing at the Universitat Autònoma de Barcelona are financing the research of this disease in our group.