Predicting the Electronic Absorption Band Shape of Azobenzene Photoswitches
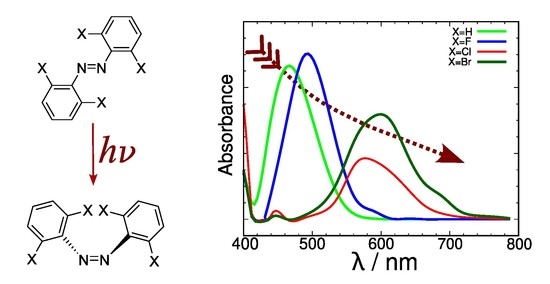
Simulations based on molecular dynamics coupled to excitation energy calculations were used to generate simulated absorption spectra for a family of halide derivatives of azobenzene, a family of photoswitch molecules with a weak absorption band around 400–600 nm and potential uses in living tissue. This is a case where using the conventional approach in theoretical spectroscopy (estimation of absorption maxima based on the vertical transition from the potential energy minimum on the ground electronic state) does not provide valid results that explain how the observed band shape extends towards the low energy region of the spectrum. The method affords a reasonable description of the main features of the low-energy UV-Vis spectra of these compounds. A bathochromic trend was detected linked to the size of the halide atom. Analysis of the excitation reveals a correlation between the energy of the molecular orbital where excitation starts and the energy of the highest occupied atomic orbital of the free halide atom. This was put to the test with a new brominated compound with good results. The energy level of the highest occupied orbital on the free halide was identified as a key factor that strongly affects the energy gap in the photoswitch. This opens the way for the design of bathochromically shifted variants of the photoswitch with possible applications.